Inflammation is a response of a tissue to injury, often injury caused by invading pathogens. It is characterized by
- increased blood flow to the tissue causing
- increased temperature,
- redness,
- swelling, and
- pain.
A bacterial infection initiates inflammation through several interconnecting mechanisms:
- The "nonself" surface of bacteria allows the complement system to be activated through the "alternative pathway".
- Certain surface molecules of the bacteria, called Pathogen-Associated Molecular Patterns (PAMPs), bind to Toll-like receptors (TLRs) on or in a variety of leukocytes.
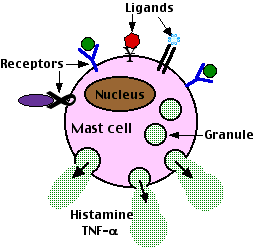
Mast cells are found in the tissues.
- Their cytoplasm is loaded with granules containing mediators of inflammation.
- Their surface is coated with a variety of receptors which, when engaged by the appropriate ligand, trigger exocytosis of the granules.
Mast cells appear to be key players in the initiation of inflammation.
- Their Toll-like receptors trigger exocytosis when they interact with PAMPs like
- Their receptors for complement fragments trigger exocytosis when they bind
Activated mast cells release literally dozens of potent mediators;
- some immediately as they discharge their granules
- some later as they synthesize them by new gene transcription.
These mediators are active in either (or, in some cases, both)
- recruiting all the types of white blood cell to the site
- activating many of these recruited cells to produce their own mediators of inflammation.
I shall not attempt to catalog all the players, but here are some of the major (and best understood) ones.
Large amounts of TNF-α are quickly released by stimulated mast cells. All the cells involved in inflammation have receptors for TNF-α and are activated by it to synthesize more on their own. This positive feedback quickly amplifies the response.
Tryptase is the most abundant protein released by mast cells. It is a serine protease. Like the mammalian enzyme trypsin, tryptase cleaves peptide bonds on the C-terminal side of arginines and lysines. It activates C3 of the complement system and probably supports inflammation in other ways as well.
These are chemotactic cytokines; that is, secreted proteins that attract other leukocytes into the area. Several have been identified.
These are produced by activated phagocytes: macrophages and neutrophils. They are toxic for microorganisms but can also lead to tissue injury. ROS are described in detail on another page. Link to it.
The granules of mast cells are loaded with histamine and their exocytosis releases this potent mediator. Histamine increases the blood flow to the area and the leakage of fluid and proteins from the blood into the tissue space. Thus the quick release of histamine produces the redness and swelling associated with inflammation.
Macrophages, monocytes, and activated platelets are sources of this cytokine. IL-1 has both
- paracrine effects on cells in the vicinity, e.g.,
- causing them to produce tissue factor and thus triggering the blood clotting cascade. [Link]
- stimulating the synthesis and secretion of a variety of other interleukins
- helping to activate T cells and thus initiate an adaptive immune response
- endocrine (hormonal) effects as it is carried in the blood throughout the body.
- decreasing blood pressure
- inducing fever.
IL-1 causes fever by stimulating the release of prostaglandins, which act on the temperature control center of the hypothalamus.
Inflammasomes
IL-1 is synthesized from a larger precursor that is cleaved by a caspase (caspase-1). Caspase-1 is part of a multi-protein complex in the cytosol of macrophages and neutrophils called an inflammasome. Inflammasomes are activated by several different products produced by invading bacteria. Some of these are first "seen" by toll-like receptors (TLRs) thus providing a link between the innate immune system and inflammation.
Bradykinin is a nonapeptide (9 amino acids).
It is synthesized by proteolytic cleavage of an inactive precursor (a kininogen) that is produced by the liver and circulates at all times in the blood (one of the alpha-globulins).
Bradykinin
- relaxes the smooth muscle walls of the arterioles lowering blood pressure and increasing blood flow to the tissue and
- makes the capillaries leakier, allowing blood components to enter the tissue space.
- These effects (like those of histamine) produce the redness, warmth, and swelling of inflammation.
The process:
- Hageman factor (also known as clotting factor XII [12]) normally circulates in the blood as inactive precursor.
- When tissue damage allows blood to escape into the tissue space, Hageman factor comes in contact with the collagens in the tissue space and becomes activated.
- Activated Hageman factor is a serine protease that cleaves an inactive precursor called prekallikrein into another serine protease — kallikrein.
- Kallikrein then cleaves kininogen forming bradykinin.
Bradykinin also
- stimulates the release of nitric oxide;
- stimulates phospholipase to increase the production of prostaglandins and
- (mediates the closing of the ductus arteriosus when a baby is born).
- plays a major role in the dangerous swelling associated with hereditary angioedema (HAE) — [Link].
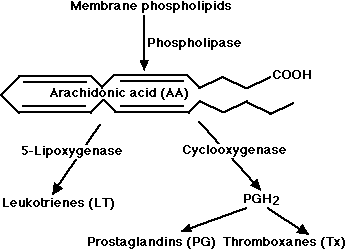
These potent mediators of inflammation are derivatives of arachidonic acid (AA) a 20-carbon unsaturated fatty acid produced from membrane phospholipids.
The principal pathways of arachidonic acid metabolism are
- the cyclooxygenase (COX) pathway, which produces prostaglandin H2 (PGH2). PGH2 serves as the substrate for two enzymatic pathways: one leading to the production of several
- prostaglandins (PG); the other leading to the production of
- thromboxane (Tx).
- the 5-lipoxygenase pathway, which produces a collection of leukotrienes (LT)
The acute inflammatory response to tissue damage is of great value. By
- isolating the damaged area,
- mobilizing effector cells and molecules to the site, and — in the late stages —
- promoting healing,
inflammation protects the body.
Its importance is demonstrated by the problems people with inherited defects in components of the process have with infections.
Some examples:
- a failure to produce reactive oxygen species (ROS) leads to chronic granulomatous disease (CGD) [Link to discussion]
- inherited defects in the ability to produce the later complement components (C5, C6, C7, C8, C9) increase the risk of certain infections.
In chronic inflammation, the inflammatory response is out of proportion to the threat it is faced with or is directed against inappropriate targets. In the first case, the result can be more damage to the body than the agent itself would have produced.
- All the many types of allergies and
- many of the autoimmune diseases
are examples of inflammation in response to what should have been a harmless agent.
Some examples:
In many of these cases, the problem is made worse by the formation of antibodies against
- self antigens or
- persistent antigens from smoldering infections.
The antibodies complex with the antigens triggering the complement system with all its mediators of inflammation.
The result: immune complex disorders.
Inappropriate inflammation can be treated with
- steroids like the glucocorticoid cortisol
- nonsteroidal anti-inflammatory drugs (NSAIDs) like aspirin and ibuprofen (e.g., Motrin®, Advil®).
- a number of therapeutic proteins produced by recombinant DNA technology.
The NSAIDs achieve their effects by blocking the activity of cyclooxygenases.
The body produces two different forms of cyclooxygenase:
- COX-1, which is involved in pain, promoting clotting, and protecting the stomach;
- COX-2, which is involved in the pain produced by inflammation.
In addition to reducing the fever and pain of inflammation, NSAIDs also inhibit clotting. They do this by interfering with the synthesis of thromboxane A2 in platelets. This is the reason that
- aspirin is given to patients undergoing angioplasty;
- many people take a baby aspirin a day in the hope of avoiding heart attacks.
But regular use of NSAIDs has a downside: a tendency to develop ulcers in the stomach and duodenum.
Most of the NSAIDs inhibit both COX-1 and COX-2. However, some newer drugs, the so-called COX-2 inhibitors, such as
- rofecoxib (Vioxx®)
- celecoxib (Celebrex®)
are much more active against COX-2 than COX-1.
COX-2 inhibitors are effective against inflammation and avoid damage to the GI tract. But, unfortunately, they increase the risk of blood clots — which can cause heart attacks and strokes — because they do not block the synthesis of thromboxane A2 by platelets (which contain only COX-1). So people depending on NSAIDs for their heart protective effects must monitor any use of COX-2 inhibitors carefully.
In fact, because of the increased risk of heart attacks and strokes, the manufacturer of Vioxx® removed it from the market on 30 September 2004.
Recombinant DNA and monoclonal antibody technology have produced some new therapies that are being enlisted in the battle against damaging inflammation.
- an IL-1 antagonist that binds and inactivates the IL-1 receptor.
- etanercept (Embrel®). A soluble version of the TNF-α receptor. It binds TNF-α preventing it from carrying out its many inflammatory actions. Potent but carries a severe risk of allowing infections to develop.
- recombinant protein C. To help the body dissolve the tiny clots that are triggered during inflammation.
- Infliximab (Remicade®). Binds to tumor necrosis factor-alpha (TNF-α). Shows promise against some inflammatory diseases such as rheumatoid arthritis (by blunting the activity of Th1 cells). Side-effects: can convert a latent case of tuberculosis into active disease; can induce the formation of autoantibodies (by promoting the development of Th2 cells).
In fact, all the more powerful anti-inflammatory agents (e.g., glucocorticoids) increase the risk of infection.
On occasions, for reasons that are not entirely clear, the inflammatory response — usually to an infection by lipopolysaccharide (LPS)-bearing Gram-negative bacteria — spirals out of control progressing until it involves the entire body. This life-threatening development is called sepsis.
The circulatory system loses its integrity:
- There is a breakdown of the adherens junctions between the cells lining the capillaries allowing fluid to leak into the tissue spaces — edema.
- There is a breakdown in the control of blood clotting. What should have been a mechanism to help wall off an infected area and promote healing leads instead to a dangerous deposition of fibrin in small blood vessels throughout the body.
If these responses are massive, they can lead to septic shock
- a failure of many organs: lungs, kidneys, etc.
- a sharp drop in blood pressure and, all too often,
- death
Some Gram-positive cocci can produce a similar condition, but here the eliciting agent is not LPS but a toxin liberated by the bacteria.
In theory, anti-inflammatory agents should be useful in combating sepsis. But so far, only recombinant protein C has shown any promise (by inhibiting the formation of thrombin), and severe bleeding is a dangerous side-effect.
Chronic inflammation is also a frequent cause of cancer.
- Liver cancer is often the sequel to years of inflammation caused by infection by hepatitis B and/or C viruses.
- Lung cancer often is the end stage of years of chronic inflammation caused by inhaled irritants, of which tobacco smoke is the most reliable.
- Cervical cancer can follow chronic infection and inflammation caused by
- Chronic infection with the liver fluke Opisthorchis viverrini is responsible for many cases of bile duct cancer in Thailand and Laos.
- Bladder, colon, pancreas, stomach, and other cancers may similarly be the final stage of years of inflammation.
The strong link between chronic inflammation and cancer should not be surprising when you consider that
- the reactive oxygen species (ROS) liberated during inflammation are powerful DNA-damaging agents [Link];
- increased mitosis in response to inflammation puts more cells at risk of mutations as they replicate their DNA during S phase;
- But the effect of inflammation in causing cancer may be quite independent of its effect on DNA. There is a growing body of evidence that humans harbor many cells that have accumulated enough "driver" mutations to become cancers but do not. Those that do may have done so because of their exposure to the soup of inflammatory molecules liberated by the many types of cells that gather at sites of inflammation. So a cell with enough mutated oncogenes to become a cancer may nonetheless remain dormant until triggered by chemicals in its surroundings.
- Apoptosis, the programmed death of damaged cells, is suppressed in inflamed tissue. So cells with precancerous genetic mutations, which should have committed suicide, live on to grow into a full-blown cancer.
18 January 2024