The most common hemoglobin of adult humans is known as hemoglobin A (HbA). However, other variant forms of hemoglobin are found in some humans. One of these is called hemoglobin S (HbS). Individuals who manufacture HbS exclusively suffer from sickle-cell disease.
Sickle-cell disease is quite common in malaria-ridden parts of Africa and Asia and also occurs in African-Americans and others descended from inhabitants of those regions.
|
|
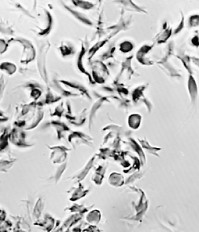
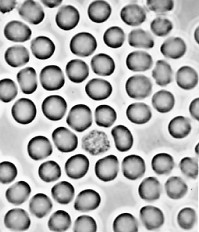
| Red blood cells of a patient with sickle-cell disease. Left: oxygenated. Right: deoxygenated. The shape of the cells when deoxygenated causes them to break easily. (Courtesy of Dr. Anthony C. Allison.) |
The disease gets its name from the fact that the patient's red cells become crescent or sickle-shaped while passing through the capillaries, especially in tissues that are metabolizing actively. The distorted cells are very fragile and are apt to rupture long before their normal life span (about 120 days) is over. This causes a severe anemia (giving rise to an alternate name for the disease: sickle-cell anemia).
Individuals with sickle-cell disease have inherited from each parent a gene — βS — encoding the beta chain of hemoglobin. Individuals who inherit only one βS gene along with the βA allele have both HbA and HbS in their red cells. In the malaria-free United States, these heterozygotes are well. In regions where malaria is common, having one of each beta chain gene (βA and βS) confers resistance to one of the most dangerous types of malaria (falciparum). This would explain why the HbS gene is so prevalent in those regions. [Link]
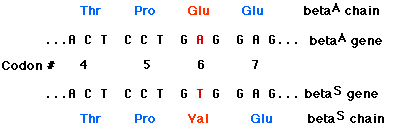
The amino acid sequences of the beta chains of HbA and HbS have been determined. The beta chains are identical except for the amino acid at position 6 (counting, as always, from the amino terminal). This position is occupied by glutamic acid in HbA chains, but in HbS beta chains, valine is found there instead.
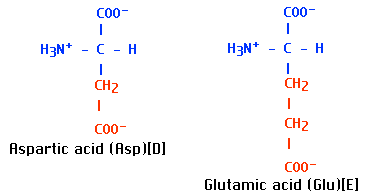
Why does this single amino acid change in a chain of 146 amino acids so drastically alter the properties of deoxygenated hemoglobin? In switching from glutamic acid to valine, a strongly hydrophilic molecule has been replaced by a strongly hydrophobic one. Position 6 is located at the surface of the beta chain, where it would normally be exposed to water. This switch from a hydrophilic to a hydrophobic region on the surface reduces the solubility of the molecule and promotes the formation of large insoluble aggregates.
The mutation that produces HbS is a single-base substitution in which the substitution of a T for an A at the 17th nucleotide of the sense strand of the first exon of the beta chain gene converts a codon for glutamic acid (GAG) to a codon for valine (GTG). Although this change might at first appear trivial, the resulting substitution of valine for glutamic acid so alters the physical properties of hemoglobin that a serious disease is produced in people carrying both genes for the trait.
| Link to a discussion of how genetic testing can reveal whether unaffected parents carry a copy of the βS gene |
| Link to a discussion of how gene therapy can provide long-term relief from the harmful features of the disease. |
| Welcome&Next Search |
{kind=link}
{kind=link}