{kind=link}
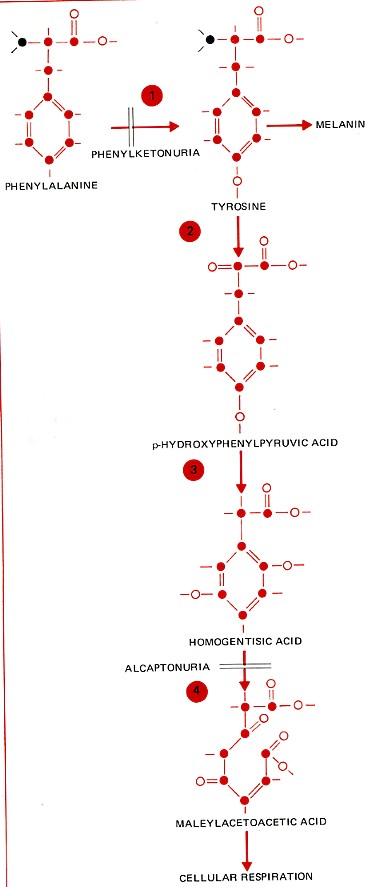
Phenylketonuria is one of the commonest inherited disorders — occurring in approximately 1 in 10,000 babies born in the U. S. It occurs in babies who inherit two mutant genes for the enzyme phenylalanine hydroxylase (PAH — "1" in the figure on the left). This enzyme normally starts the process of breaking down molecules of the amino acid phenylalanine that are in excess of the body's needs for protein synthesis.
The complete pathway is shown in the figure on the left. Phenylalanine that is in excess of the body's needs for protein synthesis is broken down as shown here and used in cellular respiration and to synthesize melanin as needed. Carbon atoms are shown in color, nitrogen atoms in black, and hydrogen atoms as short dashes.
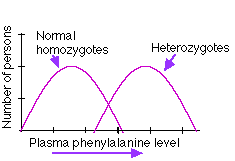
Because we inherit two copies of the gene for the enzyme, both must be defective to produce the disease. A laboratory test that measures how quickly an injection of phenylalanine is removed from the blood can distinguish a person who has one PKU gene from a person who has none, but the person with one is perfectly healthy because the unmutated allele produces enough of the enzyme. However, these heterozygous individuals are "carriers" of the disease.
| The phenylalanine tolerance test. A short time after administering a measured amount of phenylalanine to the subject, the concentration of phenylalanine in the blood plasma is measured. The level is usually substantially higher in people who carry one PKU gene (even though they show no signs of disease) than in individuals who are homozygous for the unmutated gene. Both parents must be heterozygous (i.e., must be "carriers" of the trait) to produce a child with PKU. The chance of their doing so is 1 in 4. | |
Inability to remove excess phenylalanine from the blood during infancy and early childhood produces a variety of problems including mental retardation. Fortunately, a simple test (needing only a drop of blood) done shortly after birth can identify the genetic defect and, with close attention to the amount of phenylalanine in their diet, the children can develop normally.
Alcaptonuria is another inherited disease involving this pathway. It results from the inheritance of two mutant genes for the enzyme ("4") that converts homogentisic acid to maleylacetoacetic acid. When step 4 is blocked, homogentisic acid accumulates in the blood. The kidney excretes this excess in the urine, and oxidation of homogentisic acid by the air turns the urine black. Diseases like PKU and alcaptonuria are called "inborn errors of metabolism" because they are inherited and each is characterized by a distinct metabolic defect. The condition is often spelled alkaptonuria (AKU).
In the United States, approximately 1 person in 50 has inherited a PKU allele. This means that some 5 million people in the U.S. are "carriers". Should they be tested before they decide to become parents?
By testing the DNA of prospective parents, their genotype can be determined and their odds of producing an afflicted child calculated. In the case of PKU, if both parents are heterozygous for the gene, there is a 1 in 4 chance that they will produce a child with the disease.
Problems:| Genetic screening for "a" (NOTE: not "the" — many different mutations in the PKU gene have been identified) PKU allele.
Top: schematic of a portion of
Middle: The daughter (solid circle) with PKU inherited one PKU allele from each of her parents (half-filled symbols). Her brother (open square) beat the odds (0.5) of inheriting at least one PKU allele and thus of being a carrier. If he had been a carrier, would he have wanted to know? Bottom: The blot shows the fragments to which the probe binds directly beneath each individual in the pedigree. It reveals only the 3.3 kb fragment for the brother, only the 4.2 kb fragment for the sister, and both for each parent. The particular mutation in this family is only one of many mutant PAH alleles that cause PKU, and testing with this particular enzyme and probe would not necessarily detect the others. (Based on the work of Avigad, S., et. al., Nature, 344:168, 1990.) |
 |
| Description of the process |
The disease PKU is clearly inherited as a recessive trait. Only if one inherits a mutant allele from each parent will one develop the disease.
However, heterozygous people are easily distinguished from homozygotes by the phenylalanine tolerance test. So using the test as the criterion, the PKU allele shows partial dominance.
So, the relationship between genotype and phenotype is not always straightforward. What is the criterion of phenotype in this case? the disease? the results of the tolerance test?
| External Link |
| More on screening babies for PKU |
| Please let me know by e-mail if you find a broken link in my pages.) |
| Welcome&Next Search |